By popular request, or rather the sheer number of hits the Parallelizing Freesurfer post, today we turn our attention to Tracula, the Freesurfer integrated solution for doing diffusion tensor imaging (DTI) tractography! Tracula has a lot of nice features, one of my favorites is the ability to estimate 18 or so tracts in the brain by constraining the tractography to the underlying anatomy. According to the website it uses probabilistic tractography (using FSL under the hood) with anatomical priors. You probably care about more details than this, and you probably care enough to head over to the Tracula main page and read about it there!
Of course if you haven’t already run the full Freesurfer recon-all process on your structural data, you might wish to start doing that now… Done? Great!
As with just about any kind of MRI processing, Tracula can be slow. As of Freesurfer version 5.3, I’m seeing times around 18 hours per subject on big chunky Mac Pros. Add that in with another 20 hours to run Freesurfer on all data going into Tracula and putting your data through the full pipeline can seem like quite an endeavor! But never fear, it turns out it’s fairly easy to automate Tracula (documented using their config files) and even parallelize it.
Now we all come from different approaches, Tracula documentation is setup for DICOM files as the inputs, but you can really give it anything that Freesurfer’s mri_convert will accept. So NIFTI is a perfectly valid file format to give it and might make your life easier since dcm2nii (part of MRIcron) will automatically spit out the relevant bvec and bval files that you need!
Tracula can do considerable amounts of preprocessing of your diffusion data! But, since Tracula accepts NIFTI files, that means we can (if we so desire) do quite a bit of the preprocessing (e.g. eddy current correction, blip-up/blip-down) outside of Tracula. I’ve posted before about using TORTOISE to do these things (part 1, part 2, part 3) as well as using DTIprep (tutorial, automating) as well as a general review of other DTI-related articles I’ve posted.
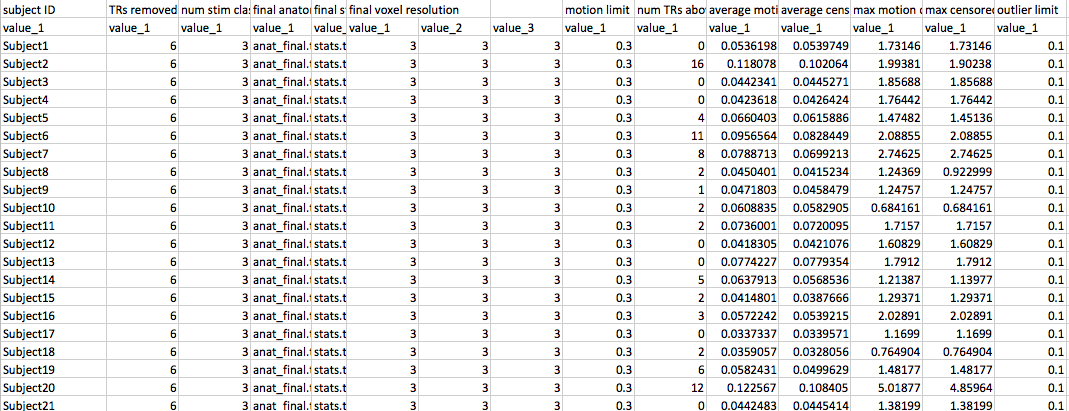
In the example here I’ve used my DTIprep script from before to do a quick run through of my diffusion run. I can then copy the output three files dwi_QCed.nii dwi_QCed.bvec and dwi_QCed.bval to whatever directory I wish to use as my input directory in Tracula. So at this point my file tree might look something like this:
-Subject001
--dwi_QCed.nii
--dwi_QCed.bvec
--dwi_QCed.bval
-Subject002
--dwi_QCed.nii
--dwi_QCed.bvec
--dwi_QCed.bval
I usually put my Tracula files in an “input” directory and this is separate (or sometimes nested within) my Freesurfer SUBJECTS_DIR. In this example I nested it within my Freesurfer folder. The next step is to setup your Tracula config file, there are great resources (read: examples) available on their official wiki. But for what I need, you might end up with something like what is shown below. Notice that because I’ve already preprocessed my data with DTIprep, I’m turning off some of the functionality in the pipeline since we probably don’t need to eddy_correct again:
#!/bin/tcsh
setenv SUBJECTS_DIR /data/freesurfer
set dtroot=/data/freesurfer/tracula
set subjlist = ( Subject001 )
set dcmroot = /data/freesurfer/tracula/inputs
set dcmlist = ( Subject001/dwi_QCed.nii )
set bvecfile = /data/freesurfer/tracula/inputs/Subject001/dwi_QCed.bvec
set bvalfile = /data/freesurfer/tracula/inputs/Subject001/dwi_QCed.bval
#don't do these, handled by DTIprep (or TORTOISE)
set doeddy = 0
set dorotbvecs = 0
#register via Freesurfer bbregister
set doregflt = 0
set doregbbr = 1
#put in MNI space
set doregmni = 1
set doregcvs = 0
set ncpts = (6 6 5 5 5 5 7 5 5 5 5 5 4 4 5 5 5 5)
Now this config file is great if you just want to run a single participant’s data through the pipeline. The config file allows you to specify more than one participant, but for our automation process here let’s leave it at one because we’re going to use the power of UNIX to make life easier! With this config file, you could execute Tracula with one command:
trac-all -prep -c NameOfConfigFile.txt
If we wanted to run this config file on another participant we could go through and change the subject number and run it again and so on and so forth. But that seems really tedious! So instead let’s use sed to change subject name and then execute the next participant:
cp NameOfConfigFile.txt NameOfConfigFile_Subject002.txt
sed -i '' "s/Subject001/Subject002/g" NameOfConfigFile_Subject002.txt
trac-all -prep -c NameOfConfigFile_Subject002.txt
Now take it one step further and run this through a loop, make your script smart enough to not re-run the script on participants (though I think Tracula may be smart enough to avoid this):
for aSub in Subject???
do
#check to see if they already exist!
if [ ! -e $current/$aSub ]; then
echo "Starting process for $aSub"
cp Tracula_Config.txt $aSub.config.txt
sed -i '' "s/Subject001/${aSub}/g" $aSub.config.txt
trac-all -prep -c $aSub.config.txt
fi
done
Now your computer will sequentially run Tracula on each participant. If you wanted to get even more crazy and parallelize the process (hence the name of the post), you could spit the Tracula command into a file and then use GNU parallel to run all jobs (here up to 8) simultaneously:
echo "trac-all -prep -c $aSub.config.txt" > all_jobs.txt
parallel -j 8 < all_jobs.txt
And there you have it. You can now easily automate Tracula and even parallelize it without having to deal with config files over and over again! To maximize your time you might want to add in lines for doing the -bedp -path and -stat options as well!